Localización de proteínas de levadura clasificadas con la tecnología de aprendizaje profundo TruAI™
Puesta a prueba de la inteligencia artificial (IA)
La tecnología de la inteligencia artificial (IA) está ayudando a aliviar la carga manual de los investigadores que tienen que procesar elevados volúmenes de datos provenientes del procesamiento de imágenes en microscopía. Una red neuronal formada por el aprendizaje profundo TruAI™ permite la segmentación
automática de objetos a partir de complejos conjuntos de datos; ¿pero qué tan adaptable y eficiente es? A través de esta nota de aplicación, se corrobora el rendimiento de la tecnología TruAI en una aplicación particularmente difícil: la clasificación dentro del cribado de alto contenido para localizar proteínas en levaduras.
¿Cuál es el desafío? El cribado de fluorescencia de alto contenido para clasificar levaduras
Conocer la localización subcelular de las proteínas es un requisito previo y clave para comprender su función biológica. Para poder examinar dónde se encuentran las proteínas en el organismo modelo Saccharomyces cerevisiae (en adelante, levadura), los científicos de este ensayo han desarrollado colecciones variadas de mutantes en el genoma. Dichos mutantes contienen proteínas que pueden llevar un marcador fluorescente en el terminal N [1,2] o C [3,4]. Esto permite a los investigadores observar la localización de la proteína mutante bajo el microscopio y estudiar su patrón fluorescente (Figura 1).
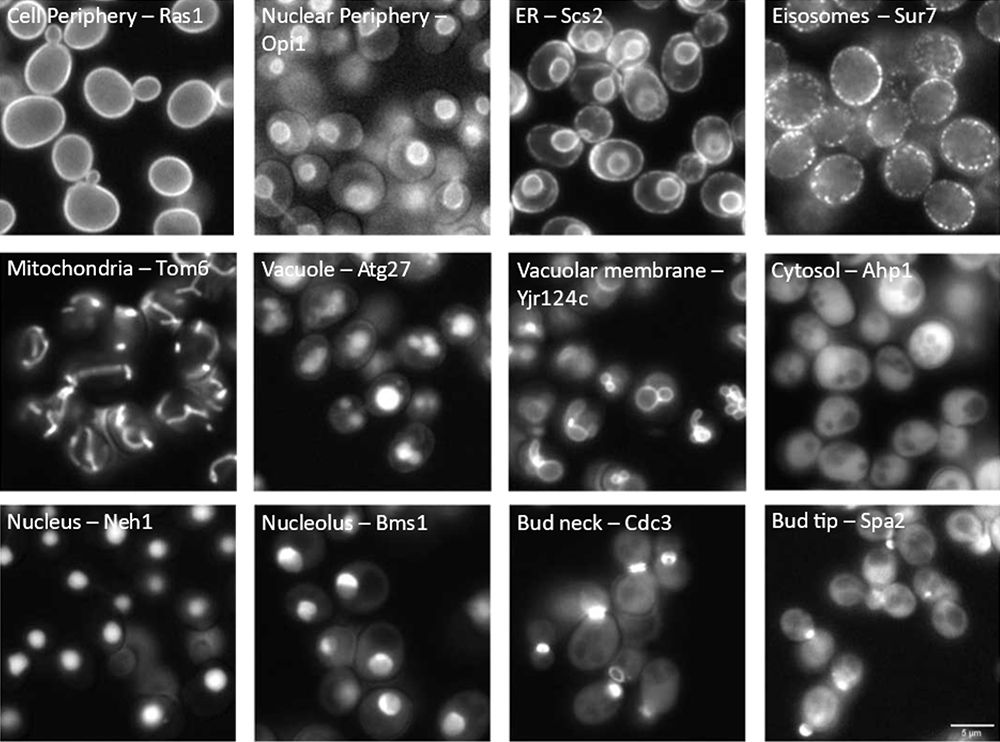
Figura 1: Localización del compartimento celular de proteínas con marcado fluorescente en la levadura. El compartimento visualizado (izquierda) y el nombre estándar de cada proteína marcada (derecha) se marcan en cada imagen.
Las levaduras tienen en total unos 6000 genes. Por lo tanto, estas colecciones de mutantes de todo el genoma requieren unas 6000 cepas mutantes individuales. Se han desarrollado tratamientos de alto contenido que permiten la manipulación genética simultánea de miles de cepas de levaduras para crear colecciones personalizadas en función de criterios científicos específicos (p. ej., al introducir una deleción de genes en una colección de mutantes fluorescentes en todo el genoma o al introducir un segundo marcador fluorescente para evaluar las relaciones espaciales entre diferentes proteínas. Hay tratamiento racionalizados y disponibles para el manejo eficiente de colecciones de mutantes extensas y para la adquisición de imágenes mediante microscopía automatizada [5]. No obstante, un obstáculo en estos ensayos de alto contenido es analizar las grandes cantidades de datos de imágenes generados con el fin de determinar la localización de proteínas en función del patrón fluorescente.
En esta nota de aplicación, se expone el uso de la tecnología de aprendizaje profundo TruAI™ en combinación con la plataforma de cribado de alto contenido scanR a fin de crear un modelo de IA basado en el patrón de fluorescencia, que permita clasificar automáticamente la localización de proteínas en diferentes compartimentos celulares a nivel de las diferentes cepas de levadura (Figura 1).
Anotaciones aprobadas en terreno y formación de la inteligencia artificial
Al desarrollar cualquier modelo de inteligencia artificial, el primer paso es crear un modelo aprobado en terreno que relaciona el patrón de píxeles específico de una imagen a una clase específica. En el caso de las tareas de segmentación de imágenes estándar, esto puede lograrse fácilmente mediante anotaciones manuales que hacen uso de las herramientas de marcado del software [6]. Sin embargo, cuantas más clases tenga que diferenciar el modelo, más anotaciones aprobadas en terreno son necesarias, lo que causa más esfuerzo al usuario y anotaciones manuales tanto ineficientes como tediosas.
La tarea deviene más compleja cuando el objetivo es crear un modelo que pueda generalizarse de forma eficaz en una amplia gama de cepas y pueda adaptarse a las variaciones acarreadas por las condiciones del procesamiento de imágenes. Estas variaciones abarcan aspectos como la calidad del enfoque, el contraste de la fluorescencia, la relación entre señal-ruido, etc., que deben tenerse en cuenta adecuadamente en las anotaciones.
Para afrontar este reto, hemos empleado un método avanzado de preparación de muestra junto con el software de cribado de alto contenido scanR, que integra a la perfección las herramientas TruAI. Este software hace más eficiente la asignación automatizada de las anotaciones aprobadas en terreno al simplificar considerablemente el proceso.
Se usó una placa de 384 pocillos para preparar y procesar en imagen diferentes cepas mutantes que expresan las proteínas marcadas con fluorescencia y cuya localización era conocida. Se seleccionaron varios representantes a partir de un total de 12 localizaciones: periferia celular, periferia nuclear, retículo endoplasmático (RE), eisosomas, mitocondrias, vacuola, membrana de la vacuola, citosol, núcleo, nucléolo, cuello de gema (o yema), punta de gema (yema). Para generar la variabilidad de fenotipos en cada clase de localización, se seleccionaron diferentes cepas independientes para cada localización hasta obtener un total de 133 cepas usadas para la formación (Figura 2).

Figura 2: Diseño de la placa de 384 pocillos dedicada a la preparación de la muestra aprobada en terreno. Cada pocillo corresponde a una cepa en la que se ha marcado una proteína específica con GFP en su terminal N. Todas las cepas de la misma fila comparten la misma localización de la proteína y se asignaron a la aprobación en terreno de la misma clase.
El procesamiento de imágenes se llevó a cabo con un microscopio de campo amplio scanR y un objetivo en aire 40x (A. N. de 0,95). Para poder identificar células de levadura individuales, se ejecutó la segmentación en el canal de transmisión mediante un modelo de inteligencia artificial preformado e integrado en el software [7] (Figura 3). Los objetos resultantes se filtraron mediante un factor de circularidad y un área para excluir artefactos y células anormales. Todas las máscaras de segmentación fueron almacenadas automáticamente en un archivo individual, compuesto por la información de los parámetros determinados a cada levadura segmentada, como el pocillo al que pertenece (es decir, la clase y la cepa) o si se han filtrado como células normales o anormales. Este archivo se utiliza en la interfaz TruAI del software scanR para crear anotaciones aprobadas en terreno para las 12 clases de células normales. Todos los píxeles filtrados como células anormales se ignoraron en la formación. De esta forma, se llegó a un número de 4 000 a 15 000 anotaciones individuales por cada clase, que engloban diferentes cepas y una serie de variaciones típicas de las imágenes (enfoque, contraste de la imagen, intensidad de la señal, fragmentos de células, etc.).
Tras haber finalizado con las segmentaciones y haber asignado la aprobación en terreno, se determinó la configuración de la formación TruAI. Se seleccionaron las opciones «Red generalizada» (Generalizing Network) y «Segmentación semántica» (Semantic Segmentation), se habilitó superposición de la clase de píxeles y formamos 350 000 repeticiones.
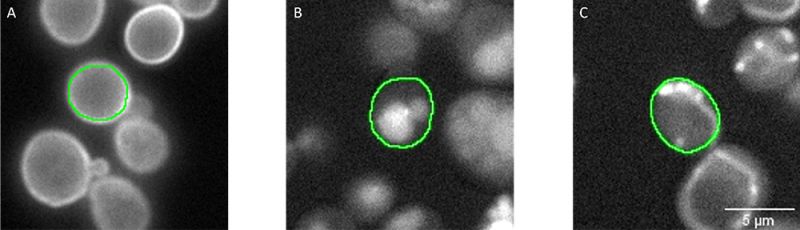
Figura 3: Máscaras de segmentación (verde) de células individuales segmentadas en el canal de transmisión (no se muestra). Las señales fluorescentes representan proteínas localizadas en A) la periferia celular (pocillo 60, C12), B) la vacuola (pocillo 266, L2) y C) la mitocondria (pocillo 147, G3). Las anotaciones aprobadas en terreno fueron asignadas automáticamente al software scanR al combinar la máscara de segmentación, el canal
fluorescente y el número de pocillo.
Resultados y validación de la solución de clasificación basada en la inteligencia artificial
Con el fin de evaluar su rendimiento real, el modelo fue evaluado mediante un conjunto de datos independientes que no habían sido incluidos en la formación. Se preparó una nueva placa de 384 pocillos con cepas expresando proteínas marcadas con fluorescencia, pertenecientes a las 12 clases de localización proteica. Tras haber aplicado el procesamiento de imágenes en transmisión y fluorescencia, se ejecutó un análisis automatizado con el software scanR en el que se aplicaron dos modelos de inteligencia artificial: un modelo de inteligencia artificial preformado e integrado para la detección de células en transmisión, y nuestro nuevo modelo de clasificación para las localizaciones de proteínas en función del patrón de fluorescencia. A fin de visualizar rápidamente los resultados del rendimiento, se usaron mapas térmicos que pueden crearse en el software y muestran el porcentaje celular a partir de cada pocillo con una elevada probabilidad de una clase específica; asimismo, se generaron galerías a partir de estas células individuales, tal como se ejemplifica en la identificación de proteínas que se localizan en los nucléolos (Figura 4).
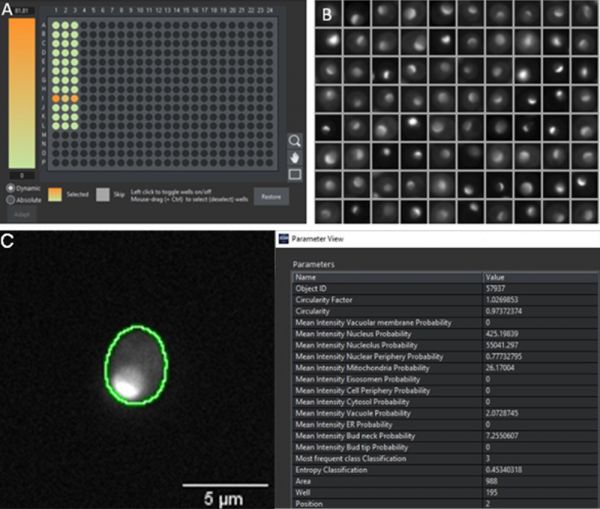
Figura 4: A) Mapa térmico aplicado a una placa de 384 pocillos que muestra la probabilidad de pertenencia de las células del pocillo a la clase de nucléolo. El mapa térmico indica que las proteínas solo se localizan en el nucléolo de la fila I. B) Galería de objetos segmentados con alta probabilidad de nucléolos destinada a confirmar visualmente un patrón de fluorescencia que corresponde con una localización del nucléolo (compárese con la Figura 1). C) Célula segmentada en el pocillo I3 dotada de un conjunto de parámetros extraídos desde la máscara de segmentación. En este ejemplo, el nucléolo presenta el valor más alto (55041), el cual es 100 veces superior al de la segunda clase de clasificación más alta (núcleo, valor 425).
Con el fin de lograr una evaluación de rendimiento más precisa para el modelo, se comparó la predicción de la clasificación de la inteligencia artificial con las anotaciones aprobadas en terreno, descritas en una matriz de confusión (Figura 5).
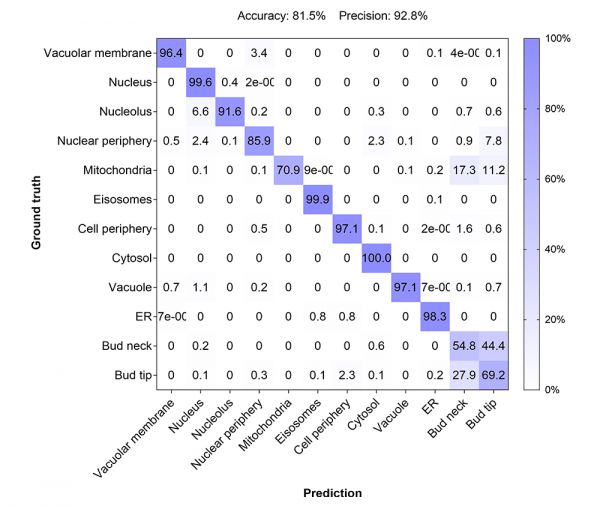
Figura 5: Matriz de confusión destinada a evaluar el rendimiento del modelo de inteligencia artificial formado en función de las clases previstas con las clases reales (de 1 600 a 4 000 células individuales por clase).
La matriz presentó una exactitud global del 81,5 % y una precisión del 92,8 % definidas como:
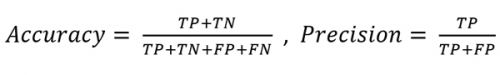 |
VP: Verdaderos positivos
|
El modelo demostró un rendimiento sólido para todas las localizaciones de proteínas excepto en el caso de los cuellos de gemas y puntas de gemas. Si bien el modelo predijo que la localización estaba en el cuello de la gema o en la punta de la gema, tuvo dificultades para distinguir entre las dos localizaciones. Este limitación podría tener una base biológica, ya que las proteínas de las dos clases demuestran una superposición considerable en la
localización en función de la fase del ciclo celular (ver Figura 1).
Conclusión: Ventajas de la localización de proteínas de levadura en el cribado de alto contenido por inteligencia artificial
Se demostró que con la preparación de las muestras y la asignación automatizada inteligentes de un modelo aprobado en terreno para miles de células, fue posible desarrollar un modelo de aprendizaje profundo por IA capaz de predecir con exactitud 10 clases diferentes de localizaciones de proteínas de levadura. Este método puede aplicarse a otras formaciones complejas de clasificación, ya que ofrece las siguientes ventajas: 1) no precisa que el usuario sea capaz
de programar un software; 2) ahorra tiempo al evitar tediosas anotaciones manuales; y, 3) gracias a la ventajosa anotación aprobada en terreno, se obtiene la capacidad de producir redes sólidas de clasificación por inteligencia artificial dedicadas a las variaciones en el procesamiento de imágenes, lo que las hace adecuadas para una aplicación por lote a muchas muestras, como las aplicaciones de cribado de alto contenido.
Referencias
- Yofe, I. et al. (2016) One library to make them all: streamlining the creation of yeast libraries via a SWAp-Tag strategy. Nat. Methods 13, 371–378
- Weill, U. et al. (2018) Genome-wide SWAp-Tag yeast libraries for proteome exploration. Nat. Methods 15, 617–622
- Huh, W.-K. et al. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–91
- Meurer, M. et al. (2018) Genome-wide C-SWAT library for high-throughput yeast genome tagging. Nat. Methods 15, 598–600
- Cohen, Y. y Schuldiner, M. (2011) Advanced methods for high-throughput microscopy screening of genetically modified yeast libraries. Methods Mol. Biol. 781, 127–59
- https://www.olympus-lifescience.com/applications/rapid-automated-detection-and-segmentation-of-glomeruli-using-self-learning-ai-technology/
- https://www.olympus-lifescience.com/discovery/20-examples-of-effortless-nucleus-and-cell-segmentation-using-pretrained-deep-learning-models/
Autores de la universidad de Münster:
Julian Schmidt, Sarah Weischer, Mike Wälte, Jens Wendt, Thomas Zobel y Maria Bohnert
Autor de Evident:
Manoel Veiga, especialista de aplicaciones, Evident Technology Center Europe
Productos usados para esta aplicación
Sorry, this page is not
available in your country.