Specimen Preparation Using Synthetic Fluorophores and Immunofluorescence
Triple-Staining Tissue Cryosections with
Wheat Germ Agglutinin, Phalloidin, and Nuclear Dyes
A majority of the common tissue sections derived from laboratory animals, including intestine, kidney, testes, muscle, liver, and the lungs, produce brightly colored fluorescent specimens detailing a wide variety of anatomical features when stained with a cocktail that includes wheat germ agglutinin and phalloidin (or phallacidin) conjugated to common low molecular weight synthetic fluorescent probes. Among the useful fluorescent markers for visualization of tissues are rhodamine, fluorescein, the Alexa Fluor series, and the cyanine dyes. Counterstaining for nuclei using a variety of popular DNA-binding dyes follows treatment with the lectin and actin probes. This protocol details a generalized procedure for staining tissue cryosections ranging from 5 to 20 micrometers in thickness.
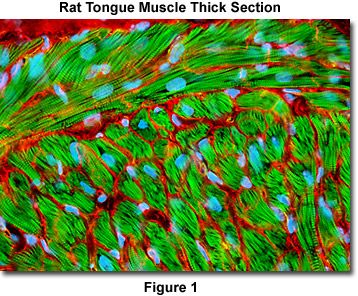
Presented in Figure 1 is a confocal image revealing striated actin fiber bundle structure in a thick (16 micrometer) section of rat tongue. The section was stained as detailed below using wheat germ agglutinin conjugated to Alexa Fluor 568 and phalloidin conjugated to Oregon Green 488. Nuclei were counterstained with Hoechst 33342. The specimen was imaged with a 60x oil immersion objective (without zoom) using a 405-nanometer violet diode laser (Hoechst), a 488-nanometer argon-ion laser (Oregon Green 488), and a 543-nanometer helium-neon laser. Images were collected in grayscale and subsequently pseudocolored with hues approximating the fluorescence emission spectra of the respective probes.
Reagents
- Fixative - 3.7 percent paraformaldehyde in PBSA (prepare fresh fixer each day).
- Permeabilization Buffer - 0.2 percent Triton X-100 in PBSA (sonicate detergent buffer for 30 minutes to one hour immediately before use).
- Blocking Buffer - 1 percent bovine serum albumen (BSA) in PBSA containing 0.05 percent Triton X-100 (add 2-3 milligrams sodium azide per 100 milliliters of blocking buffer to eliminate the growth of microorganisms).
- PBSA Wash Buffer - Use PBSA before permeabilization (after fixation) and after treatment with the phalloidin/lectin probes except for those tissues requiring specialized buffers for nuclear dyes that are not compatible with PBSA.
- PBSA-Triton Wash Buffer - For wash sequences immediately after permeabilization and after blocking (before staining with nuclear dyes), use PBSA with 0.05 percent Triton X-100.
- Wheat Germ Agglutinin/Phalloidin Cocktail - Add 10 microliters of WGA stock solution (1 milligram per milliliter) and 20 microliters of phalloidin stock solution (6.6 micromolar) to 1 milliliter of BSA Blocking Buffer. Each thin section receives 250-500 microliters of staining cocktail.
- Nuclear Dye - Prepare fresh dilutions of the nuclear dye immediately prior to staining.
- Nuclear Dye Wash Buffer - Hoechst and SYTOX dyes require Hanks Balanced Salt Solution (Hanks BSS), while DAPI, as well as the monomeric and dimeric cyanine nuclear stains, can be used with PBSA.
Nuclear Counterstain Dilutions
- Hoechst (33342 and 33258) - 5 microliters in 150 milliliters of Hanks BSS (30 minutes).
- SYTOX Green and Orange - 10 microliters in 250 milliliters of Hanks BSS (30 minutes).
- DAPI - 5 microliters in 150 milliliters of PBSA (5 minutes).
Procedure
Carefully insert the frozen and mounted cryosections (on 1 × 3-inch microscope slides) into a vertical or horizontal staining jar. Align all sections to face in the same direction and avoid scratching the surface of the frozen tissue while loading the slides. Allow the sections to thaw in the staining jar for 20 minutes before adding the paraformaldehyde fixer. Fix 5-10 micrometer sections for 15-20 minutes and 10-20 micrometer sections for 20-30 minutes. Slowly rotate the tissue sections as they are being fixed on an orbital shaker at 5-10 revolutions per minute.
After fixation, wash the tissue sections with three changes of PBSA Wash Buffer. For thin sections (5-10 micrometers) wash for 5 minutes, and for thick sections (10-20 micrometers) wash for 10 minutes. Slowly rotate the tissue sections as they are being washed on an orbital shaker at 5-10 revolutions per minute.
Dissolve the membranes in the tissue sections with Permeabilization Buffer by treatment for one or two hours for thin (5-10 micrometer) and thick (10-20 micrometer) sections, respectively. Slowly rotate the tissue sections as they are being permeabilized on an orbital shaker at 5-10 revolutions per minute.
After permeabilization, wash the tissue sections with three changes of PBSA-Triton Wash Buffer. For thin sections (5-10 micrometers) wash for 5 minutes, and for thick sections (10-20 micrometers) wash for 10 minutes. Slowly rotate the tissue sections as they are being washed on an orbital shaker at 5-10 revolutions per minute.
Block nonspecific fluorophore binding sites with Blocking Buffer. For thin sections (5-10 micrometers) block for one hour, and for thick sections (10-20 micrometers) block for two hours. Slowly rotate the tissue sections as they are being blocked on an orbital shaker at 5-10 revolutions per minute.
After blocking, remove the slides from the staining jar and carefully wipe the edges and back of the glass to remove excess Blocking Buffer. Place the slides in a humidity chamber (see Figure 2) and add the staining cocktail (250 to 500 microliters per slide) to the surface of the tissue section. Cover the humidity chamber with aluminum foil to protect the fluorophores from light. Incubate the covered slides in the humidity chamber for one or two hours (thin and thick specimens, respectively) at 37 degrees.

Return the stained slides to the staining jar and wash three times with PBSA-Triton Wash Buffer. For thin sections (5-10 micrometers) wash for 5 minutes, and for thick sections (10-20 micrometers) wash for 10 minutes. Cover the staining jar with aluminum foil (to protect from light) and slowly rotate the tissue sections as they are being washed on an orbital shaker at 5-10 revolutions per minute.
For DAPI and cyanine nuclear counterstains, add the diluted dye in PBSA to the staining jar and treat the specimen for the recommended time: 5-10 minutes for DAPI; 15-30 minutes for cyanine dyes (protect from light with aluminum foil). When using Hoechst or SYTOX stains (30 minute incubation), first wash the slides in Hanks Balanced Salt Solution for two buffer exchanges prior to counterstaining.
Wash the counterstained specimens with either PBSA or Hanks Balanced Salt Solution (depending upon the nuclear dye) for three times at 5 minutes for each wash. Protect from light with aluminum foil.
After the final washing step, remove the slides from the staining jar and wipe excess buffer from the rear surface and edges of the slide. Immediately mount the stained tissue with the selected mounting medium (do not allow the tissue to dry).
Sorry, this page is not
available in your country.